Bleeding Disorders Due To Deficiency Of Coagulation Factors: Hemophilia A (Factor VIII deficiency)

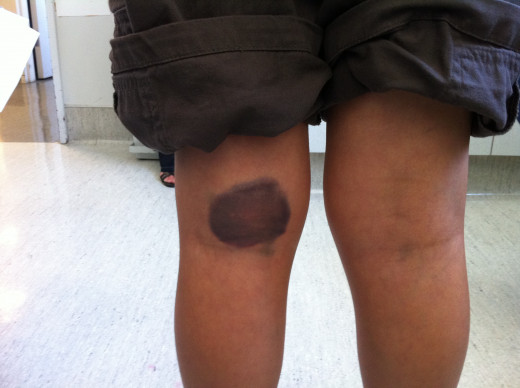
Hemophilia Bruises
Hemophilia Overview
Hemophilia is the commonest among the inherited coagulation defects. The disease is worldwide in distribution. In India for instance, it forms two thirds of all the inherited coagulation defects. About 25% of cases may not give any family history and it is presumed that in them the disease arises due to spontaneous mutation either in the patient or his mother. The clinical picture varies widely in affected subjects.
Hemophilia is transmitted as a sex-linked recessive disorder and the genetic defects is located in the X-chromosome. It is transmitted by females but it manifests almost exclusively in males. Half of the daughters of female carriers possess the trait and half of the sons suffer from the disease. Male hemophilics pass the trait to all their daughters but their sons are not affected. The basic defect is deficiency or absence of factor VIII: C (anti-hemophilic globulin (AHG)). When the level of factor VIII:C falls below 30% of normal, bleeding tendencies start. Spontaneous bleeding occurs only when the level falls below 5%. In a severe case, there may be no detectable factor VIII:C episodes of bleeding.
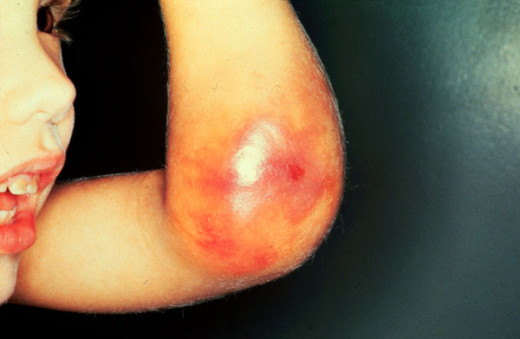
Hemophilia Manifestation
Factor VIII
It is a protein of large molecular size (1.5- 2X106 daltons) and it contains two functional components- factor VIII related antigen (VIII:R.AG) and VIII coagulant (VIII.C). Factor VIII:R.AG is larger and forms the major bulk and it is precipitated with heterologous antisera developed in experimental animals. It contains the Von Willebrand’s factor (VIII.vWF) which is concerned with platelet adhesion to exposed subendothelial connective tissue and ristocetin induced aggregation. The particular molecular configuration of factor VIII. R.AG is probably responsible for its effects on platelets. Factor VIII.C is loosely bound to factor VIII.R.AG and in this form it is transported in the plasma. Factor VIII.R.AG is produced by endothelial cells of blood vessels in several tissues. It is not known where factor VIII.C is synthesized or where does factor VIII.R.AG get fixed to factor VIII.C. In hemophilia factor VIII.C is reduced quantitatively or altered qualitatively, but factor VIII.R.AG is present normally. Factor VIII.R.AG takes part in coagulation. In Von Willebrand’s disease factor VIII.R.AG ( and hence vWF) is deficient though VIII.C is present. Since VIII.C cannot be properly transported or made functional without VIII.R.AG, the functions of both VIII.R.AG and VIII.C are defective in some forms of von willebrand’s disease. Factor VIII:C takes part in the intrinsic coagulation pathway by acting as a cofactor to factor IXa in the activation of factor X.
Hemophilia Symptoms
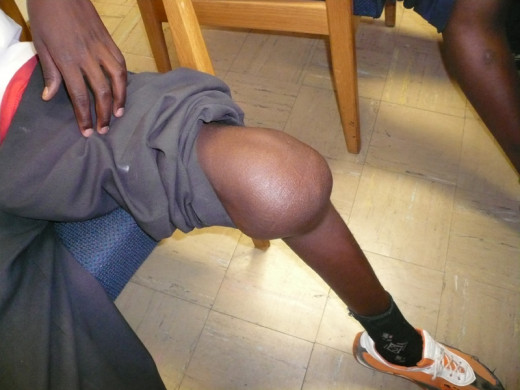
Hemophilia

Clinical Features
The bleeding tendency is noticed early in life. Sometimes, the umbilical stump may bleed. Though, bleeding can occur from any site, the common presentations are epistaxis, gum bleeding, hematomas, deep tissue bleeding, hemarthroses, or hematuria. Bleeding may be spontaneous, may follow trivial trauma or undue physical exertion. Following cuts or wounds, bleeding is arrested initially, but may start soon and proceed unchecked, leading to fatal blood loss. The extravasated blood does not clot for long periods.
Bleeding into joints occur frequently in moderate and severe cases. The knees, ankles, elbows, shoulders and hips are commonly affected. Bleeding may be spontaneous or follow unaccustomed exertion. The joints show features of acute inflammation. Repeated hemorrhage into joints and deep tissues lead to crippling ankylosis and contractures. Bleeding can occur in the central nervous system in severe cases.
The disease shows remissions and exacerbations. Physical exertion, infections, trauma, or psychological stress lead to exacerbations. With the passage of time, the severity and frequency of bleeding diminishes. Death may result from uncontrolled blood loss or bleeding into vital organs.
Diagnosis
The disease should be suspected from the history, evidence of hereditary and from the prolonged coagulation time. Bleeding time, platelet count, and prothrombin time are normal but PTTK (APTT) is prolonged. Thromboplastin generation test shows defect in the patient’s plasma and this is correctable by normal plasma. Assay of factor VIII helps in establishing the diagnosis and assess the severity. Factor VIII assay also helps to monitor replacement therapy.
Treatment Of Hemophilia
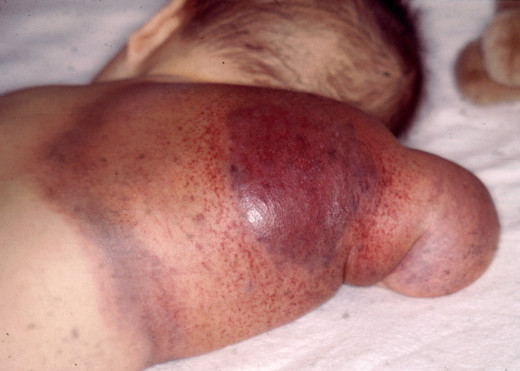
Treatment
Rest in bed is essential during a bleeding episode. The inflamed joint should be splinted and cooled with ice packs. Sedatives like diazepam or phenobarbitone or analgesics like dextropropoxyphene may be used to relieve insomnia and pain. Local measures to stop the bleeding include pressure, adrenaline packs and local dressing with fibrin-foam, thrombin or Russell viper venom. Bleeding sites from gums, tooth-sockers, or nose should be plugged with adrenaline packs. Aspiration of joint should be done only after AHG is administered. Early mobilization prevents ankylosis.
The mainstay of the treatment is administration of factor VIII concentrate. Factor VIII is available as cryoprecipitate or as freeze dried AHG. Cryoprecipitate is prepared easily from fresh frozen plasma. It contains factor VIII (both factor VIII:vWF and factor VIII:C portions). Fibrinogen and fibronectin. The dose is determined by the severity and site of bleeding. The aim is to elevate factor VIII levels to 50% or above. Cryoprecipitate is repeated every 12 hours for about two days. To prepare for major surgery and in life threatening hemorrhage, factor VIII level should be raised to 100% and cryoprecipitate has to be given every 8 hours. With improvement, the dose can be reduced to 50% and this has to be continued till bleeding stops. Since cryoprecipitate may cause urticeria and allergic reactions, prior administration of an antihistamine like phenergan is beneficial.
© 2014 Funom Theophilus Makama




